
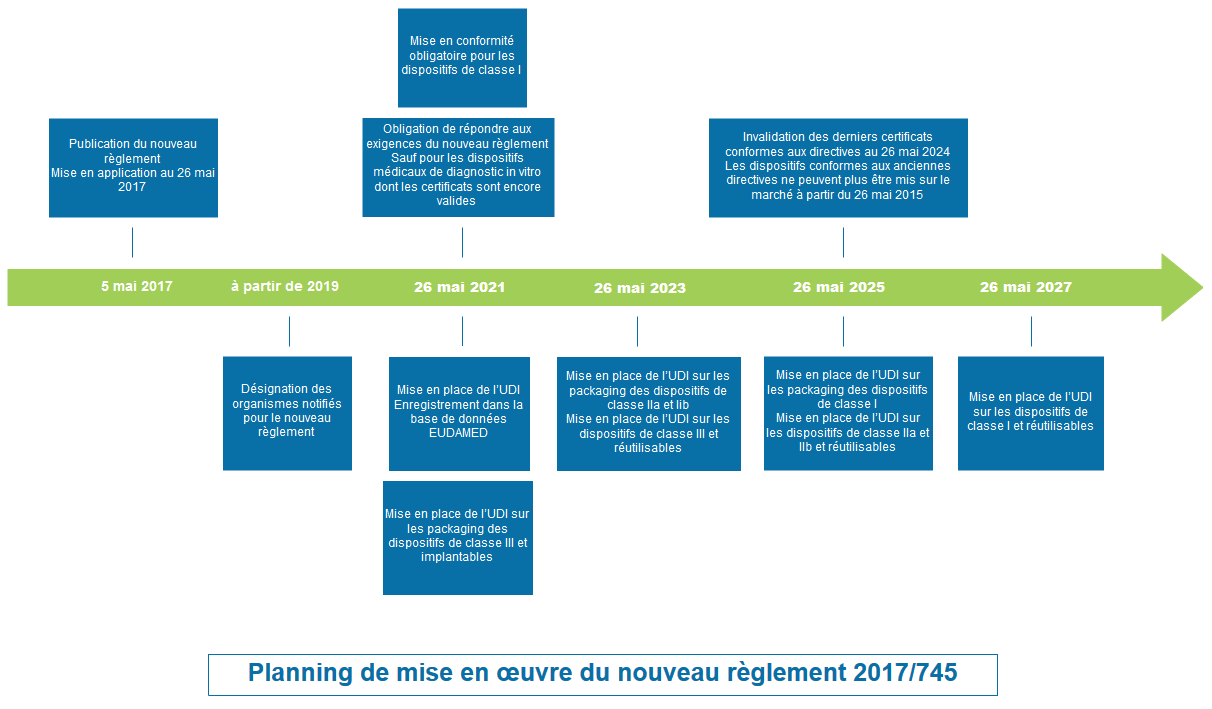
Dates de mise en œuvre (Article § 120)
- Publié le 5 mai 2017
- Entré en vigueur le 26 mai 2017
- Application obligatoire à partir du 26 mai 2020 (4 ans supplémentaires possibles pour les dispositifs médicaux déjà marqués CE à la date du 26 mai 2020 et en fonction de la date d’expiration de leur certificat).
- Les certificats délivrés par des organismes notifiés conformément aux directives 90/385 et 93/42 à partir du 25 mai 2017 conservent leur validité jusqu’à la fin de la période indiquée sur ces certificats, laquelle ne peut dépasser 5 ans. Ils sont toutefois invalidés au plus tard le 27 mai 2024
- Les certificats délivrés conformément à l’annexe IV des directives 90/385 et 93/42, sont invalidés au plus tard le 27 mai 2022
Attention, les délais sont très courts pour les nouveaux dispositifs et/ou les nouveaux fabricants.
Attention également pour les dispositifs qui vont changer de classe et dont les certificats et/ou les autodéclarations seront invalidés le 26 mai 2020.
Planning de mise en oeuvre du règlement 2017/745
- Dans ce planning, voir aussi la mise en oeuvre de l’UDI (Identification Unique du Dispositif)
- Enregistrement dans la base de données EUDAMED
- Mise en place de l’UDI sur les packaging
- Mise en place de l’UDI sur les dispositifs
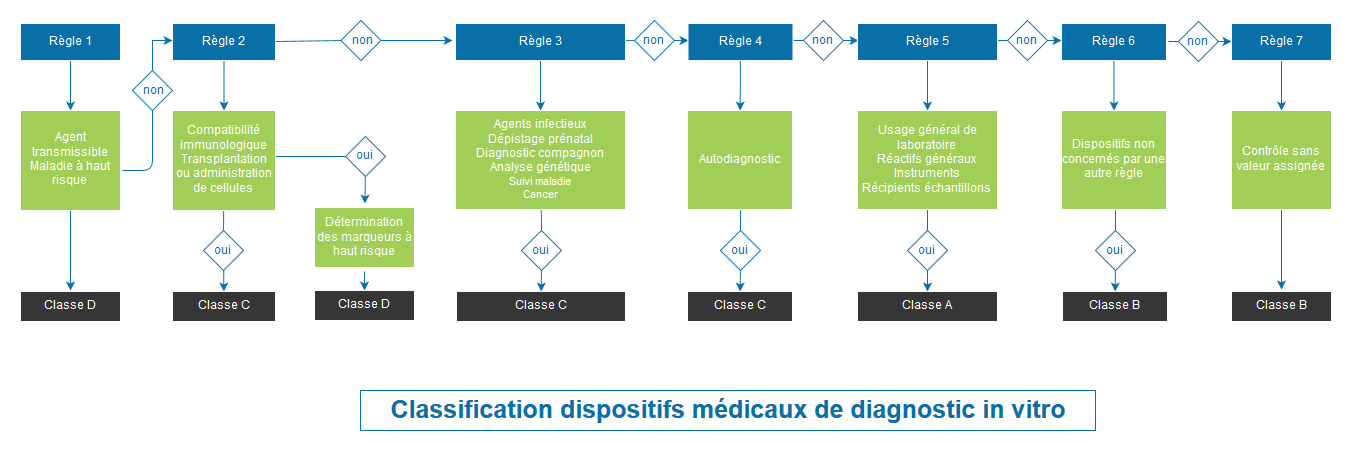
Classification des dispositifs médicaux selon le règlement 2017/745 – Annexe VIII
Nouvelles précisions:
Pour le calcul de la durée, on entend par «utilisation en continu»:
- la durée totale d’utilisation du même dispositif sans tenir compte d’une interruption temporaire d’utilisation au cours d’une procédure ou du retrait temporaire pour le nettoyage ou la désinfection du dispositif. Le caractère temporaire de l’interruption de l’utilisation ou du retrait est établi au regard de la durée de l’utilisation avant et après la période pendant laquelle l’utilisation est interrompue ou le dispositif est retiré; et
- l’utilisation accumulée d’un dispositif destiné par le fabricant à être immédiatement remplacé par un autre du même type
Un dispositif est réputé permettre un diagnostic direct lorsqu’il fournit lui-même le diagnostic de la maladie ou de l’état pathologique en question ou lorsqu’il fournit des informations décisives pour l’établissement du diagnostic
Nouvelles règles
Règle 11: Les logiciels destinés à fournir des informations utilisées pour prendre des décisions à des fins thérapeutiques ou diagnostiques relèvent de la classe IIa, sauf si ces décisions ont une incidence susceptible de causer:
- La mort ou une détérioration irréversible de l’état de santé d’une personne, auxquels cas ils relèvent de la classe III, ou
- Une grave détérioration de l’état de santé d’une personne ou une intervention chirurgicale, auxquels cas ils relèvent de la classe IIb.
Les logiciels destinés à contrôler des processus physiologiques relèvent de la classe IIa, sauf s’ils sont destinés à contrôler des paramètres physiologiques vitaux, lorsque des variations de certains de ces paramètres peuvent présenter un danger immédiat pour la vie du patient, auxquels cas ils relèvent de la classe IIb.
Tous les autres logiciels relèvent de la classe I.
Règle 19: Tous les dispositifs qui incorporent un nanomatériau ou qui en sont constitués relèvent:
- De la classe III s’ils présentent un potentiel d’exposition interne moyen ou élevé,
- De la classe IIb s’ils présentent un faible potentiel d’exposition interne,
- De la classe IIa s’ils présentent un potentiel d’exposition interne négligeable.
Règle 20: Tous les dispositifs invasifs en rapport avec les orifices du corps, autres que les dispositifs invasifs de type chirurgical, destinés à administrer des médicaments par inhalation sont de classe IIa, sauf si leur mode d’action a une incidence essentielle sur l’efficacité et la sûreté du médicament administré ou s’ils sont destinés à traiter une affection susceptible de mettre la vie en danger, auquel cas ils relèvent de la classe IIb.
Règle 21: Les dispositifs qui sont composés de substance ou de combinaisons de substances qui sont destinées à être introduites dans le corps humain par un orifice du corps ou par application sur la peau et qui sont absorbées par le corps humain ou dispersées localement dans celui‐ci relèvent:
- De la classe III si les substances en question, ou les produits de leur métabolisme, sont systématiquement absorbés par le corps humain conformément à la destination du dispositif,
- De classe III si les substances en question atteignent leur destination dans l’estomac ou plus loin dans le tractus gastro‐intestinal et si elles, ou les produits de leur métabolisme, sont systématiquement absorbés par le corps humain,
- De classe IIa si les substances en question sont appliquées sur la peau ou si elles sont appliquées dans la cavité nasale ou buccale jusqu’au pharynx et atteignent leur destination dans ces cavités, et
- De classe IIb dans tous les autres cas.
Règle 22: Les dispositifs actifs thérapeutiques ayant une fonction de diagnostic intégrée ou incorporée qui détermine largement la prise en charge du patient par le dispositif, tels que les systèmes en circuit fermé ou les défibrillateurs automatisés externes, relèvent de la classe III.